
Contenido patrocinado
Enfermedades Genéticas
“Con la XLH no hay dos pacientes iguales: es importantísima una atención multidisciplinar”
Al ser una enfermedad genética, está presente desde el nacimiento y acompaña al paciente para el resto de su vida, y, con el paso del tiempo, los síntomas se van haciendo más intensos.

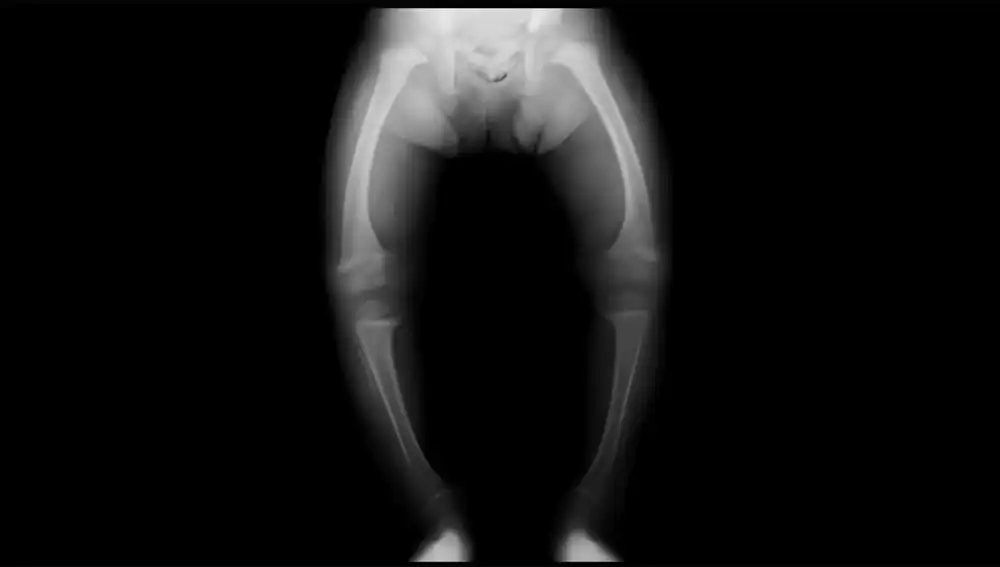
La XLH es una enfermedad rara (eso significa que es poco frecuente), en la que un defecto del gen PHEX produce una actividad excesiva y descontrolada de FGF23 (la hormona que ayuda a los riñones a eliminar el exceso de fósforo que ingerimos en la dieta), haciendo que los riñones pierdan mucho fosfato por la orina (fosfaturia), por lo que los niveles de fosfato en sangre bajan (hipofosfatemia) y no queda suficiente para mantener la salud de los huesos (formados principalmente por calcio y fósforo), entre otros procesos biológicos. “Por ello, los huesos son incapaces de mantener una estructura firme, siendo muy propensos a deformarse y romperse”, explica la Dra. Vanessa Pérez-Gómez.
La XLH es una enfermedad rara, genética, crónica y progresiva, ¿cómo afecta esta patología en la calidad de vida de adolescentes y adultos?
Esta enfermedad tiene un alto impacto psicológico y condiciona la vida en general de quienes la padecen. Para ellos es difícil el poder realizar actividades cotidianas de la vida diaria como levantarse de la cama, andar, moverse… Imagina si hablamos de otras actividades que requieran más esfuerzo como el ejercicio, actividades de ocio o ciertas profesiones. Por otro lado, deben acudir muchas veces al hospital, ya sea por los síntomas, las complicaciones o por las intervenciones quirúrgicas necesarias para tratar las deformidades.
¿Cuáles son los mayores retos a los que se enfrenta un paciente adulto con XLH?
Un paciente adulto con XLH habitualmente tiene la carga de haber sido un niño con XLH, que ha tenido disminución del crecimiento, deformidades en las piernas y alteraciones en la marcha, que han ido progresando... En la edad adulta puede presentar fracturas espontáneas con facilidad, dolores persistentes en huesos, articulaciones y músculos, abscesos dentales y pérdida de audición, pudiendo llegar a rigidez articular y debilidad muscular y fatiga generalizada. A esto se les suman las múltiples visitas a los hospitales, lo que hace que la vida con XLH sea un reto.
La XLH es una patología con gran variabilidad fenotípica, a pesar de ello, ¿cuáles consideras que podrían ser los signos y síntomas más comunes en el adulto con XLH?
El que la enfermedad exprese una amplia variabilidad fenotípica significa que no hay dos pacientes iguales, ni siquiera dentro de la misma familia: uno puede tener síntomas leves y otro, síntomas más graves. He tenido algún paciente cuya primera expresión de la enfermedad ha sido una fractura espontánea (es decir, sin golpe que la justifique) en la edad adulta y otros que, desde la infancia, cuando empiezan a dar los primeros pasos, presentan deformidades en las piernas (se arquean hacia adentro o hacia afuera porque los huesos son blandos, están poco mineralizados y no aguantan el peso del cuerpo). También en los adultos se pueden encontrar defectos en el crecimiento, osteoartritis, abscesos dentales o pérdida de audición.
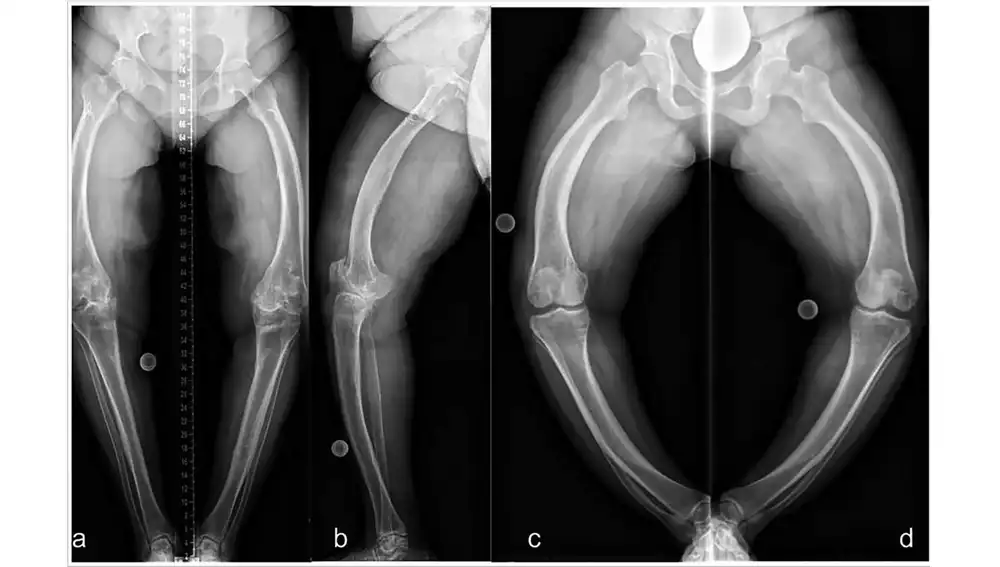
Por todo ello, es importantísimo el manejo multidisciplinar para estos pacientes, donde estamos implicados los pediatras, nefrólogos, endocrinos, reumatólogos, médicos internistas, traumatólogos, genetistas, especialistas en rehabilitación y trabajadores sociales.
Desde el punto de vista de tu especialidad en nefrología, ¿cuáles son los mayores retos a los que te enfrentas en el manejo de la XLH?
Por un lado, está el reto de alcanzar el diagnóstico en los pacientes con fosfato bajo en sangre. Primero hay que averiguar si esto está producido por aumento de las pérdidas renales de fosfato. Luego, nuestra misión es buscar entre todas las causas de pérdida renal de fosfato, cuál es la definitiva en nuestro paciente.
Por otro lado, una vez realizado el diagnóstico de XLH, el tratamiento con fosfato oral y vitamina D puede producir una entidad que llamamos nefrocalcinosis (calcificación del riñón) por el alto trasiego de fosfato desde el intestino a la sangre y a la orina, u otras complicaciones en otros órganos y a largo plazo incluso una enfermedad renal crónica.
¿Crees que es necesaria la concienciación del público general acerca de la XLH? ¿Podría esto favorecer el diagnóstico y manejo de estos pacientes?
Es una enfermedad poco común y eso implica que muchas veces pasa desapercibida, al no sospecharse y lleva a la ausencia de un diagnóstico oportuno. Un retraso en el diagnóstico resultaría en un retraso en el tratamiento. Un público concienciado podrá acudir al médico antes si observa, por ejemplo, un niño con piernas arqueadas o retraso en el crecimiento, o si es un adulto con una fractura espontánea o producida por un esfuerzo leve. En esta enfermedad, un diagnóstico precoz seguido de un tratamiento óptimo es crucial para controlar los síntomas, prevenir las complicaciones y mejorar la calidad de vida.
En la era de Internet, asistimos cada vez con mayor frecuencia a situaciones en que pacientes jóvenes con enfermedades raras se diagnostican a sí mismos, consultando sus síntomas en internet. Una mayor concienciación del púbico general puede facilitar esta vía, dado que la carrera de Medicina está enfocada a formar médicos generales y no se hace el suficiente énfasis en las enfermedades raras.
Si desea conocer más sobre la
patología, por favor acceda a
Página de acceso exclusivo
a profesionales sanitarios:
Autorretrato
Soy Nefróloga. Trabajo en la Fundación Jiménez Díaz en Madrid como facultativo especialista adjunto al servicio de Nefrología. Soy responsable de la consulta de nefropatías hereditarias y la consulta de ensayos clínicos, además de profesora asociada de la Universidad Autónoma de Madrid. En este momento coordino el grupo de trabajo de nefropatías hereditarias de la Sociedad Española de Nefrología.
Un proyecto de LR Content
✕
Accede a tu cuenta para comentar


